ABSTRACT
In this study, the adsorption mechanism and electronic properties of the CH₄ molecule on a six-layer Ni(111) surface were investigated within the framework of density
functional theory (DFT) using the GGA–PBE exchange-correlation functional implemented in the QuantumATK simulation package. The Ni(111) surface was modeled as a 4×4 surface
supercell with a sufficiently large vacuum spacing, and the geometric optimization procedure was applied to the CH₄ molecule and the top three Ni layers. The optimization
results indicate that the methane molecule preserves its ideal tetrahedral geometry (C–H ≈ 1.11 Å). The optimized C–Ni distance is approximately 3.21 Å, while only a slight
variation is observed in the Ni–Ni interatomic distance (2.49 → 2.48 Å). These structural parameters suggest that the adsorption process has a physical rather than chemical
nature. The adsorption energy calculated from total energy differences is Eₐds = +1.22 eV, indicating that adsorption in the considered configuration is thermodynamically
unfavorable, and the CH₄ molecule does not spontaneously bind to the Ni(111) surface. Analysis of the density of states (DOS) reveals that the distribution of Ni 3d
orbitals remains practically unchanged. The energy levels associated with the C and H atoms do not introduce new electronic states near the Fermi level. This confirms the
absence of significant orbital hybridization and indicates that C–H bond activation does not occur in the system.
Therefore, the obtained results demonstrate that the methane molecule interacts with the Ni(111) surface only through weak dispersion interactions, remains electronically
passive under cryogenic conditions, and does not become activated on the pristine nickel surface.
These findings are consistent with the high chemical stability of methane observed in nickel-based liquefied natural gas (LNG) infrastructures and provide a fundamental
atomic-scale explanation of processes occurring at the methane–nickel interface.
Keywords: Methane adsorption, Ni(111) surface, Density Functional Theory (DFT), Charge redistribution, liquefied natural gas (LNG).
DOI:10.70784/azip.2.2026110
Received: 02.03.2026
Internet publishing: 11.03.2026 AJP Fizika A 2026 01 az p.10-15
AUTHORS & AFFILIATIONS
1. Azerbaijan State Oil and Industry University, 20 Azadlig ave. Baku, AZ 1010, Azerbaijan
2. Institute of Physics Ministry of Science and Education Republic of Azerbaijan, 131 H.Javid ave, Baku, AZ-1073, Azerbaijan
E-mail: vusala.cafarova@asoiu.edu.az
Graphics and Images
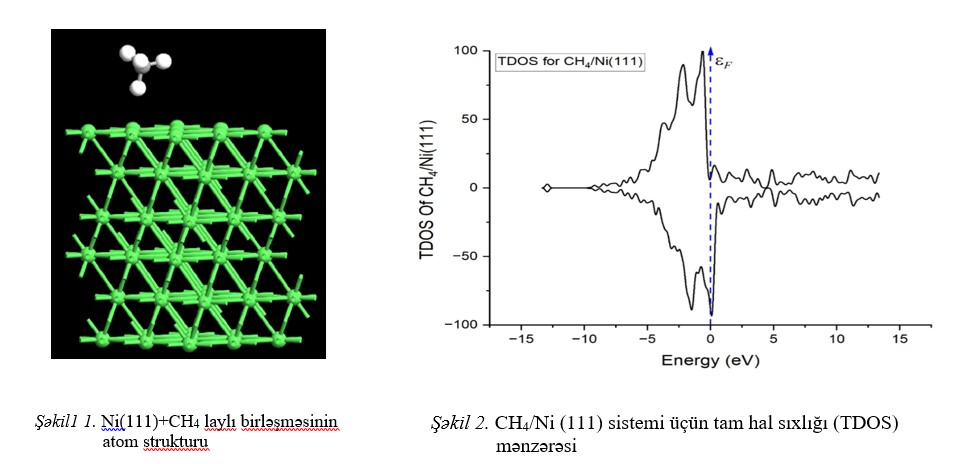
Fig.1
|
[1] S. Mokhatab, W.A. Poe, J.G. Speight. Handbook of Liquefied Natural Gas, Elsevier, Amsterdam (2014) 1–572.
[2] BP Statistical Review of World Energy, Natural Gas Overview, BP Energy Economics (2023)1–70.
[3] Martínez, M.Y. Solís. Global outlook for LNG supply chain efficiency improvements, Energy Conversion and Management 243 (2021) 114365.
[4] P.J. Luyben. Design and control of LNG processes, Chemical Engineering Science 187 (2018) 107–118.
[5] A.R. Sadeghi, S.M. Mousavi. Thermodynamic modelling of methane under cryogenic LNG conditions, Journal of Natural Gas Science and Engineering 83 (2020) 103526.
[6] J. Sehested, J.B. Gelten, S. Helveg. Sintering of nickel catalysts under methane steam reforming conditions, Journal of Catalysis 223(2) (2004) 432–443.
[7] T. Bligaard, J.K. Nørskov, S. Dahl, J. Matthiesen, C.H. Christensen, J. Sehested. Brønsted–Evans–Polanyi relations in heterogeneous catalysis, Journal of Catalysis 224(1) (2004) 206–217.
[8] Z. Zhai, J. Lu, H. Wang, Y. Xu. Ni-based catalysts for low-temperature methane activation: DFT and experiments, Applied Catalysis B 292 (2021) 120145.
[9] S. Linic, M.A. Barteau. Mechanism of CH₄ activation on transition metal surfaces, Journal of the American Chemical Society 126(23) (2004) 8086–8087.
[10] R.L. Arevalo, R. Poloni, I. Matanović. Tuning methane decomposition on stepped Ni surfaces: Role of subsurface atoms, Scientific Reports 7 (2017) 14050.
[11] X. Niu, Y. Zhao, J. Li, A. Du. Methane activation on Ni/MgO catalysts: A DFT study, Frontiers of Chemical Science and Engineering 16(3) (2022) 403–414.
[12] M. Shirazi, G. Dehghan, E.C. Neyts. DFT study of Ni-catalyzed plasma dry reforming of methane, Journal of Molecular Catalysis A: Chemical 426 (2017) 473–482.
[13] M.T. Darby, E.C.H. Sykes, A. Michaelides. Fundamental principles of methane activation on metal surfaces, Nature Catalysis 1 (2018) 760–770.
[14] R.M. Martin. Electronic Structure: Basic Theory and Practical Methods, Cambridge University Press (2004) 1–624.
[15] J. Neugebauer, M. Scheffler. Adsorbate–substrate interactions via total-energy calculations, Physical Review B 46(24) (1992) 16067–16080.
|
